The severe acute respiratory distress syndrome coronavirus (SARS-CoV-2) enters human cells with the help of its spike protein that binds the angiotensin-converting enzyme 2 (ACE2) receptor of the host. The spike protein-host membrane reaction depends on proteolytic cleavage, as well as activation of viral envelope glycoproteins, by host cell proteases. Notable proteases in this regard include transmembrane protease, serine 2 (TMPRSS2), cathepsin L (CTSL), and cathepsin B (CTSB).
Therapies available today target either different stages of the viral life cycle (nucleotide analogs or broad-spectrum antivirals), or the host's immune system (immunosuppressive drugs or monoclonal antibodies), or vascular acute damage (antihypertensive and anticoagulant drugs). Due to their role in initiating viral infection, the ACE2 receptor and TMPRSS2 may be the most crucial therapeutic targets.
To date, antiretrovirals have been the focus of most therapeutic research, followed by anticancer drugs (e.g., kinase inhibitors) and antimicrobials as the next most promising drug candidates. Due to the high cost and time required for de novo drug development, drug repositioning is emerging as a viable option.
Traditional high-throughput screening (HTS) involves testing thousands to millions of small molecules in parallel. HTS 'hits' allow the identification of therapeutic targets and to validate biological effects even when little is known about the compound. However, it comes at a substantial cost in terms of time and resources, requiring screening of libraries of hundreds of thousands of small molecules to obtain a few (0.01% – 0.14%) active compounds for further investigation. Virtual screening can overcome these glitches in the early phases of drug development. Additionally, drug-likeness or absorption, distribution, metabolism, excretion, and toxicity (ADMET) criteria can be embedded into the process to increase the selected candidates' quality further.
Bioinformatic strategies based on data from multiple omics analyses, mechanisms of action, and different molecular alterations of coronavirus disease (COVID-19) have been proposed for drug repositioning and identifying novel drugs for COVID-19 have been proposed. On the other hand, cheminformatics strategies, based on quantitative structure-activity relationship (QSAR) modeling and molecular docking have also been employed by researchers to screen for therapeutic targets.
Researchers recently published a study in the journal Briefings in Bioinformatics wherein they integrated multiple bioinformatics and cheminformatics-based methods to prioritize drugs for the treatment of COVID-19.
Study details
The entire study framework was designed to work with four complementary bioinformatics approaches. The primary assumption behind these approaches was that topologically central genes in the network have a pivotal role in the adaptation to exposure to the virus. Consequently, researchers prioritized the drugs according to the importance of their gene targets in the network. Researchers then identified a robust rank of the drugs employing the Borda method and extracted a list of relevant chemical substructures.
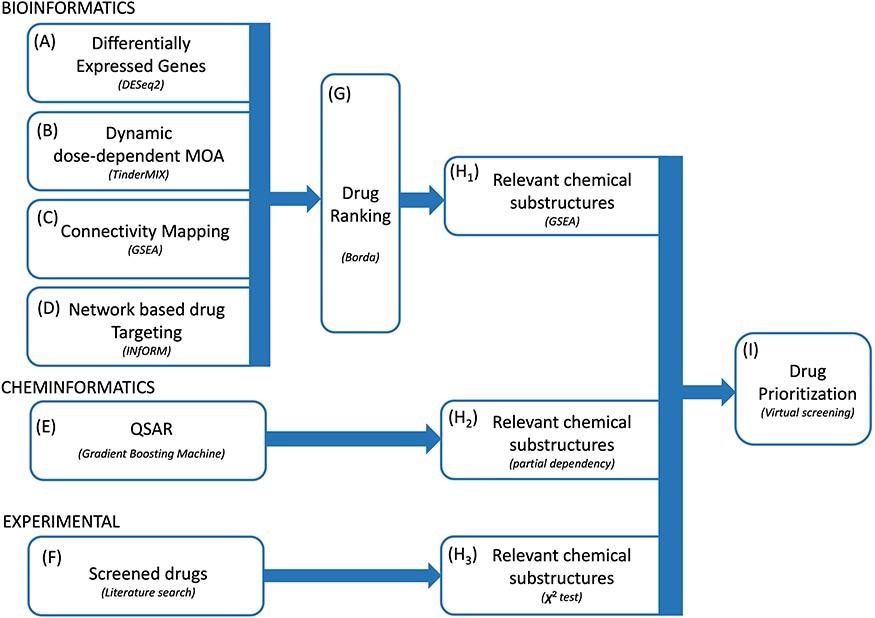
Proposed methodology. We integrated multiple bioinformatics and cheminformatics methods to prioritize drugs for the treatment of COVID-19 (A–E). Our framework consists of four complementary bioinformatics approaches, including differential expression analysis (A), dynamic dose-dependent MOA (B), connectivity mapping (C) and network-based drug targeting (D) as well as a QSAR-based cheminformatics method (E). We further complemented our set of candidate chemical substructures with those extracted from active drugs as experimentally tested in multiple studies (F). The four bioinformatics approaches are merged to find a robust rank of the drugs (G). From the rank produced by the bioinformatic approaches, the QSAR method and from the list of screened drugs, three lists of chemical substructures are identified (H1–H3) with the aim of increasing the robustness of the predictions as well as to generate knowledge readily usable in the context of de novo drug development. Eventually, we exploited the set of candidate chemical substructures by performing a virtual screening analysis of the DrugBank database (I).
On the other hand, researchers used QSAR-based cheminformatics methods to identify chemical substructures of drugs predictive of the deregulation level of the ACE2 receptor, the transmembrane protease TMPRSS2, and the cell surface proteolytic enzymes CTSB and procathepsin L (CTSL).
Based on the presence of these chemical substructures, a list of 700 candidate drugs effective against COVID-19 were identified from the DrugBank database amidst 8,000 others. The inclusion criteria were a trade-off between selecting a set of drugs that best represented the top of the prioritized list, based on their chemical substructures, and several practical considerations such as price, availability, shipping time, and ease of storage. Researchers validated their method by performing an in vitro biological evaluation of 23 selected drugs considering these criteria.
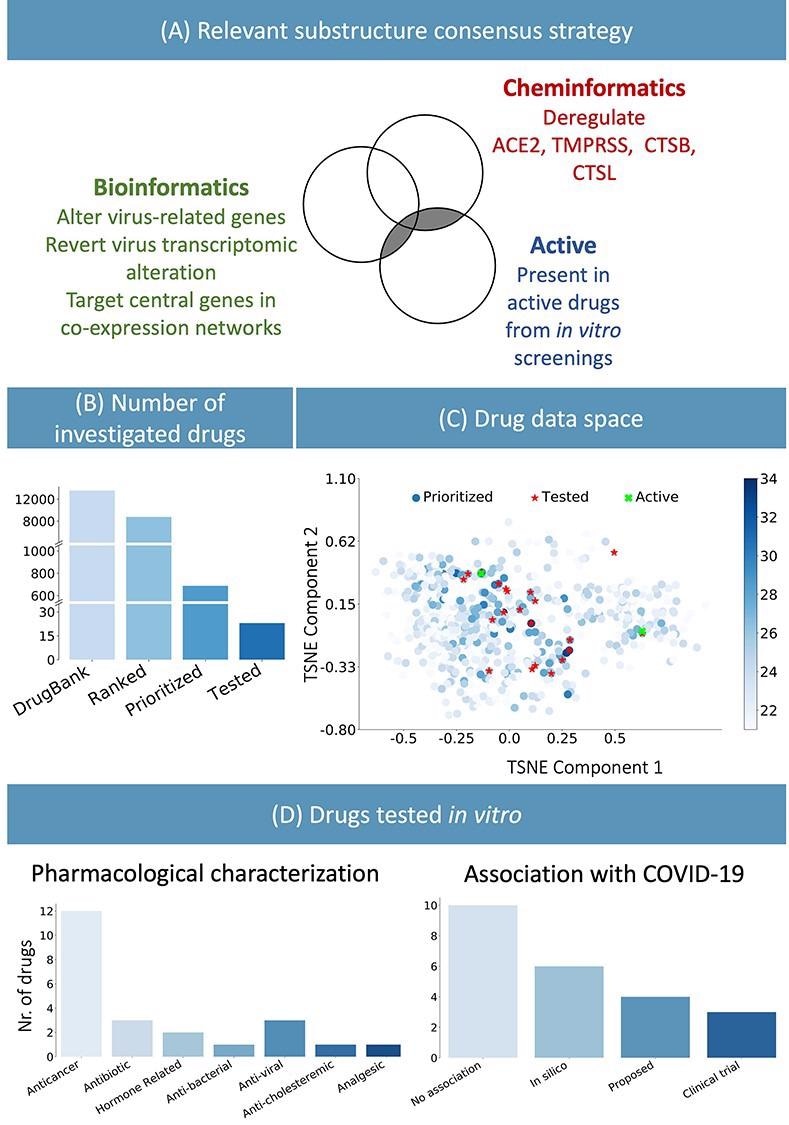
(A) Consensus strategy to identify relevant chemical substructure, using bioinformatics and cheminformatics methods as well as experimental results from published literature. (B) The suggested approach allows reducing the number of experimental tests: the whole DrugBank database was filtered to less than 800 relevant drugs and in vitro testing was performed on 23 candidates. (C) Graphical representation of the prioritized drugs. The shade blue represents the number of chemical substructures identified in (A), present in the drugs. The 23 selected compounds are shown in red. They were selected among the drugs sharing the most relevant substructure as well as satisfying practical logistic criteria. Of the 23 drugs, the two highlighted in green have been experimentally identified as active. (D) Pharmacological characterization and description of known association with COVID-19 of the 23 tested drugs. In silico refers to drugs derived from in silico studies, whereas proposed refers to drugs suggested for their potential therapeutic role in literature.
12 out of the 23 identified drugs were targeted oncology therapies, and 8 of them were kinase inhibitors. Similar to antiviral drugs, anticancer drugs may also target biological processes that play a crucial role in modulating the immune response, cell division and death, cell signaling, and microenvironment generation in the host system.
Researchers found that two drugs, 7-hydroxystaurosporine, and bafetinib, showed significant inhibition of viral infection. In addition, image analysis of the infected versus treated cells showed that the formation of multinucleated syncytial cells was also significantly reduced. Unexpectedly, when combined, the two drugs exerted an even more potent, synergistic inhibition of viral infection as well as cell-cell fusion inhibition at lower concentrations. Further in vitro experiments showed that the drugs in combination were effective even after an hour of infection of the cells. This suggested that the combination might have hindered a post-entry mechanism of the virus. Moreover, the results also confirmed the effectiveness of combining the drugs against the more infective SARS-CoV-2 delta variant.
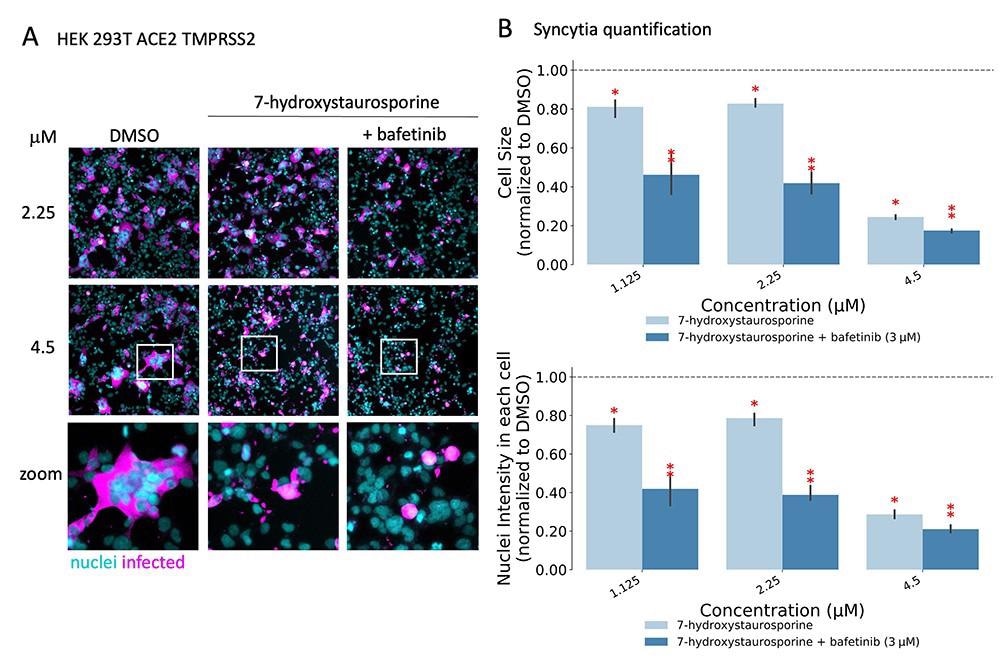
7-Hydroxystaurosporine and bafetinib inhibit virus-induced syncytia. (A) Representative fluorescence images of HEK-293 T-AT cells treated with indicated drugs 1 h before infection. Cells fixed 16 hpi; cyan = nuclei, magenta = infected cells. Zoomed areas from each image are indicated by white boxes. (B) Quantification of cell size and nuclear content from the experiment in (A); values normalized to the median of DMSO controls. All values represent the averages of three experiments. Error bars indicate the SD. Red asterisks show significant P-values (<0.05) for the one-tailed t-test between each treatment and the DMSO.
Implications
The current study demonstrated a high throughput and effective method for modeling drugs specific to COVID-19, as well as probing therapeutic targets. Furthermore, this study showed that 7-hydroxystaurosporine and bafetinib combined had the potential to effectively control SARS-CoV-2 damage. In addition to providing a first line of reference in managing emergency situations, it would also be important for future research into therapeutics against COVID-19.
- Serra A, Fratello M, Federico A, et al. Computationally prioritized drugs inhibit SARS-CoV-2 infection and syncytia formation. Brief Bioinform. 2021 Dec 27; bbab507.
Posted in: Medical Research News | Disease/Infection News | Pharmaceutical News
Tags: ACE2, Acute Respiratory Distress Syndrome, Angiotensin, Angiotensin-Converting Enzyme 2, Antibodies, Anticoagulant, Bioinformatics, Cell, Cell Division, Cell Signaling, Compound, Coronavirus, Coronavirus Disease COVID-19, Drugs, Enzyme, Fluorescence, Gene, Genes, High Throughput, High-throughput screening, Immune Response, Immune System, in vitro, Kinase, Membrane, Nucleotide, Oncology, Protein, Receptor, Research, Respiratory, SARS, SARS-CoV-2, Serine, Severe Acute Respiratory, Spike Protein, Syndrome, Therapeutics, Vascular, Virus

Written by
Sreetama Dutt
Sreetama Dutt has completed her B.Tech. in Biotechnology from SRM University in Chennai, India and holds an M.Sc. in Medical Microbiology from the University of Manchester, UK. Initially decided upon building her career in laboratory-based research, medical writing and communications happened to catch her when she least expected it. Of course, nothing is a coincidence.
Source: Read Full Article