Retinitis pigmentosa (RP) is a disease condition that was first identified and named by Dr. Donders in 1857. Retinitis pigmentosa is a group of related conditions that are inherited, progressive and clinically distinctive and share a similar feature of dystrophy or damage to the photoreceptors of the retina and of the pigment epithelium underneath the photoreceptors.

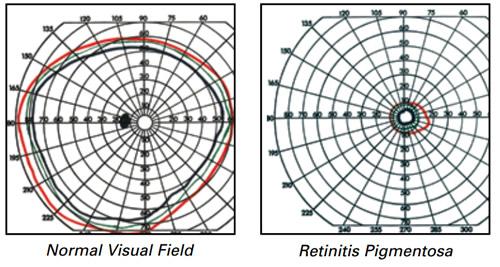
Normal retina and Retinitis pigmentosa
The normal retina is a layer or membrane that lies at the back of the eyeball. It is a photosensitive layer with millions of photo receptors that are stimulated when light rays fall on them. These photo receptors or rods and cones transmit the information of the light patterns to the brain via nerves that converge to form the optic nerve. This results in normal visual perception.
Pathophysiology and symptoms of Retinitis pigmentosa
In Retinitis pigmentosa there are initial vision problems especially in dim light. This manifests as a loss of vision around the peripheries. This is known as tunnel vision. Central vision is spared until the later stages of the disease.
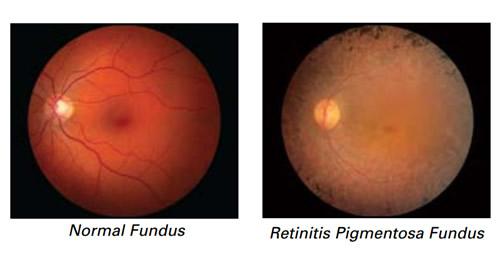
Retinitis pigmentosa is the leading cause of inherited retinal degeneration-associated blindness. The disease process begins with changes in pigment and damage to the small arteries and blood vessels that supply the retina. There may be atrophy or damage to the optic nerve as well. The damage to the pigment epithelium occurs due to pigmentation that begins and settles within the layers of the neural retina.
As the disease progresses there is a thinning of the retinal blood vessels. Gradually the light-sensitive cells of the retina deteriorate. Both rod and cone photoreceptors are affected and the type of genetic defect determines which cells are more affected. Rod photoreceptor malfunction is the most commonly encountered problem in RP.
Retinitis pigmentosa and genetics
To date, more than 50 different genetic defects have been identified to be linked with Retinitis pigmentosa. Around 30-40% are autosomal dominant, 50 to 60% are autosomal recessive and 5 to 15% are X chromosome linked (passed from mother to son and carried by daughters to their sons).
The autosomal dominant forms tend to have milder symptoms with onset usually seen at around fifth or sixth decades of life. The X-linked form is the most severe with loss of central vision seen as early as in the third decade of life itself.
Retinitis pigmentosa statistics
Retinitis pigmentosa is the leading cause of inherited retinal degeneration-associated blindness. The prevalence is 1 in 4,000 among all age groups and 1 in 3,195 persons in population aged between 45 and 64. X linked variety affects men more than women and other types have no sex or ethic preferences.
Diagnosis and treatment of Retinitis pigmentosa
Diagnosis is usually made on the basis of examination of the retina using slit lamp biomicrosopy.
There is no cure for Retinitis pigmentosa and no treatment is known to stop the progressive vision loss in the condition. There are a number of research areas including gene therapy and antioxidant therapies that are being explored in the treatment of the condition.
Sources
- www.bbc.co.uk/…/retinitispigmentosa1.shtml
- http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0002024/
- https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=791
- curriculum.usc.edu/…/retinitis_pigmentosa_case.pdf
- biochem118.stanford.edu/…/Kallini.pdf
- www.kellogg.umich.edu/…/Understand-Retinitis-Pigmentosa.pdf
Further Reading
- All Retinitis Pigmentosa Content
- Retinitis Pigmentosa Symptoms
- Retinitis Pigmentosa Diagnosis
- Retinitis Pigmentosa Genetics
- Retinitis Pigmentosa Cause
Last Updated: Apr 25, 2019

Written by
Dr. Ananya Mandal
Dr. Ananya Mandal is a doctor by profession, lecturer by vocation and a medical writer by passion. She specialized in Clinical Pharmacology after her bachelor's (MBBS). For her, health communication is not just writing complicated reviews for professionals but making medical knowledge understandable and available to the general public as well.
Source: Read Full Article